![]()
![]()
Understanding the evolutionary history of our own species, how migration and mixture of ancestral populations have shaped modern human populations is a key question in evolutionary biology. Here we present three articles related to this topic, the first two dealing with India and the third one focusing on a single Ethiopian group :
1) Moorjani et al 2013 Genetic Evidence for Recent Population Mixture in India AJHG 93,: 422–438
2) Basu et al 2016 Genomic reconstruction of the history of extant populations of India reveals five distinct ancestral components and a complex structure PNAS online before print
3) Van Dorp et al 2016 Evidence for a Common Origin of Blacksmiths and Cultivators in the Ethiopian Ari within the Last 4500 Years: Lessons for Clustering-Based Inference PLOS Genetics 11(8): e1005397
All of them use genome wide data from micro array. After a brief abstract of each paper, showing their similarities and differences, we discuss their methodological approaches.
Ancestral populations of India
The aim of the first two articles is to understand the history of the populations of the Indian subcontinent. The first one (Moorjani et al 2013) reports data from 73 groups living in India for more than 570 individuals sampled. The authors filtered out the data by removing all individuals with evidence of recent admixture or recent ancestry from out of India. The populations that were included in the analysis can be classified into two linguistic categories: the ones speaking Indo-European languages and the ones speaking Dravidian languages.

Previous genetic evidence indicates that most of the groups of India descend from a mixture of two distinct ancestral populations: Ancestral North Indians (ANI) and Ancestral South Indians (ASI). Three different hypothesis exist for the date of mixture of these two populations:
1) arrival of ANI is due to migration prior to agriculture about 30,000-40,000 years ago
2) ANI arrived with the spread of agriculture who probably began around 8,000 and 9,000 years ago
3) ANI arrived very recently (3,000-4,000 years ago) when the Indo-European languages presumably began to be spoken in India.
To prove the admixed origin of Indian groups and estimate the proportion of each ancestry in each population they use a PCA and a statistic called F4 ratio that infers the mixture proportion measuring the correlation in allele frequencies between each pair of groups. They demonstrated that all populations are admixed and lie along an “Indian cline”, that is a gradient going from 17% of ANI ancestry to 71%. These results correlate well with geography and language, with the northern Indo-European populations having more ANI ancestry than the southern Dravidian ones. Then they use linkage disequilibrium (LD) to estimate the dates of admixture : LD blocs are longer if the admixture is younger. By fitting an exponential function to the decay of LD (that is expected from a sudden cessation of admixture) they could estimate that admixture occurred between 1,856 and 4,176 years ago, supporting the third hypothesis. These results correspond with demographic and cultural changes observed in India with the establishment of the caste system leading to strong endogamy that stopped the admixture rapidly. Moreover they found that Indo-Europeans groups have more recent admixture dates, which could be explained by multiple waves of mixture in these populations. Another finding of this paper is that aboriginal Andaman Islanders (Onge) belong to a sister group of ASI.
The second article (Basu et al 2016) has the same focus region and use the same basic dataset, except that the authors kept the all populations in the analyses, including the austro asiatic (AA) and tibeto burman (TB) speakers. They first ran ADMIXTURE on all populations and showed that islanders and mainland populations have distinct ancestral components (islanders share ancestry with oceanic peoples like Papuans). In a second time they ran the same analysis on mainland populations only (thus excluding population from the Andaman and Nicobar islands). The best model was composed of four ancestral components, the ANI, the ASI as well as the ancestral AA and TB and they found that several present day populations are almost pure representatives of these ancestral components (figure 2).

They further estimated the time and extent of admixture using the degree of fragmentation (due to recombination) of haplotypes blocs originating from a donor population into the recipient population. In each population, the distribution fitted again with an exponential curve. They showed that admixture abruptly came to an end about 1575 years ago in upper-caste populations, most likely due to the establishment of endogamy, while tribal populations seemed to have admixed until 1500-1000 years ago.
In short, although they share a common topic, these two papers propose divergent versions of the history of Indian population : while the first considers a priori that austro asiatic and tibeto burman speakers are not component of the ancestral populations of India and only focuses on the mixture between the ANI and ASI components, the second paper claims that the genetic structure of Indian population is the result of admixture events between four ancestral components. However the two views converge on the idea that admixture was a common phenomenon in India that ceased rapidly with the establishment of the caste systems that enforced endogamy.
Common origin of two subgroups of Ari people
The 3rd paper investigates the history of human populations at a smaller scale, focusing on a single ethnic group, the Ari people of Ethiopia. The Ari are composed of two socially and genetically distinct subgroups : the cultivators (Aric) and the blacksmiths (Arib). Anthropologists have proposed two alternatives hypothesis to explain the division of the Ari : under the remnant hypothesis (RN), the blacksmiths are the remnants of an indigenous group that was assimilated by the more recently arrived cultivators, whereas the marginalization (MA) hypothesis proposes that the two groups share a common ancestry but the blacksmith were recently marginalized due to their activity. While anthropologists traditionally favour the MA hypothesis, recent genetic studies have provided support for the RN hypothesis. In this article the authors use a new methodology on the same genetic dataset to bring evidence for the MA hypothesis. They show that when ADMIXTURE, fineSTRUCTURE or CHROMOPAINTER analysis are run on a complete dataset of 237 samples of 12 Ethiopian and neighbouring populations, the Arib are grouped into a single homogeneous cluster. But when the patterns of haplotype sharing are inferred by composing the Ari as a genetic mixture of all other groups, except themselves, the genetic differences between Arib and Aric disappear. In fact, their analyses reveal that the two Ari groups have the same mixture events with non Ari populations (figure 3).

To explain this pattern they propose that the genetic differentiation of the blacksmith is due to a bottleneck effect. Their hypothesis is supported by the fact that identity-by-descent (IBD) is stronger in blacksmiths than cultivators which is consistent with reduced genetic diversity in the blacksmiths. Using the D-statistic, they also show that the Arib and Aric are more closely related to each other than they are to any other Ethiopian group. Therefore they conclude that the observed genetic differentiation between the Arib and Aric does not represent separate ancestry but is rather the result of strong genetic drift due to a bottleneck effect induced by the social marginalization of the blacksmiths.
Methodological discussion
What stands out from reading these three articles is that selection of a proper methodology is crucial within an hypothesis testing framework. While the two articles on Indian populations use the same initial dataset, the way they filter and analyse it results in very different conclusions. The inclusion or exclusion of some populations from an admixture analysis or outgroup selection for an f4 ratio estimation directly impact the output of these analysis and can lead the authors to tell very different stories. Before disclaiming or putting forward one hypothesis, it is important to be aware of the limitations of the method that is used to produce the results. For example the authors of the second paper on India’s ancestral populations, claim to demonstrate a more complex history than shown in the first paper but their result is solely based on a clustering analyse (implemented in various softwares such as STRUCTURE or ADMIXTURE).
The basic principle of those STRUCTURE/ADMIXTURE like programs is to take the K most different groups of the dataset, consider them as the pure ancestral groups and force the others to be a combination of those. This means that the results depend on the populations and the number of clusters K that are input in the program. There are different methods to determine which K provide the best fit to the data (cross-validation error, delta K …) but in numerous cases the inferred mixture proportions are wrong. Only in very simple cases, like the African American genetic history (well explained in Daniel Falush’s blog) that involves three clearly defined and very differentiated ancestral populations (West Africans, Europeans and Native Americans) we can be confident in the results of the clustering analyse.

But in many cases the history is more complex and no current population actually corresponds to a pure ancestral population because of multiple waves of admixtures. In this case the most differentiated groups correspond only to the most extreme groups but it does not mean that these groups are pure or ancestral. This is well explained in Razib Khan’s blog using the simple example of Uygurs and Europeans : it is known that the Uygurs are a recently mixed group (between European and Asian) but if K is fixed to 2 with Uygurs and Europeans, STRUCTURE will form two different clusters at 100% levels, one with the Uygurs and one with Europeans. This is why, in the 2nd paper, the apparently pure AAA, ATB, ASI and ANI populations and all the clustering implications are probably meaningless. In fact, when using the f4 ratio (as in the first paper) all groups are found to be admixed to a certain extent (with the smallest rate of admixture being 17%).
This critic of clustering analysis is a key element of the study on the Ari people where the authors point out that results from such methods should not be taken for granted but interpreted with caution. Indeed this kind of method cannot discriminate between alternative scenarios of recent mixture of separate populations or shared ancestry followed by population divergence. Therefore support for one of these hypotheses should rely on additional tests. Instead of directly accepting the story suggested by a clustering analysis, a more reasonable work-flow would be to use other methods in order to address the specific implications of one hypothesis. This is exactly what is done in the third article where, as we previously explained, the authors constrain the analysis of mixture by forbidding self ancestry in the two groups of interest which remove the confounding effect of recent bottleneck. In such complex cases, associating PCA and STRUCTURE-like analyses with F-statistics and simulations allow to draw a more robust conclusion. Indeed statistics such as Fst or Dxy that estimate the genetic differentiation between two populations can be simulated under alternative scenarios, representing competing hypothesis (figure 5). These simulated statistics can be subsequently compared with the ones estimated from real data to favour one hypothesis over the other. Simulations can also give an idea of how difficult it is to discriminate between the different hypothesis, which avoid over interpretation of the results. In the second paper, where the authors put forward an new hypothesis, radically different from the classical hypothesis of anthropology and other genetic studies, additional tests like these seem necessary to strengthen their conclusions.

Although it was not mentioned in any of the articles, the quality of the data and the way to obtain them, i.e. the kind of sequencing methodology, should also be a matter of precaution. Indeed, they all use micro arrays designed from European populations. These micro arrays consist of thousands of DNA spots containing a predefined sequence, known to be polymorphic in Europeans and only the complementary sequence can fix to this spot and be sequenced. So using these micro arrays to study the history of non european populations may be problematic as only SNPs that are variable for europeans will be targeted, probably leading to the exclusion of meaningful information for non European populations. Today, with New Generation Sequencing (NGS) there are many alternatives, such as RAD sequencing or Whole Genome Sequencing, that allow to sequence tens of thousands non-predefined SNPs.
Conclusion
To conclude, the take home messages from these three articles are :
– Social systems leading to endogamy can influence and modify rapidly and dramatically the genetic structure and patterns of humans populations.
– It is difficult to reconstruct the ancestry of human populations, especially when they involve a complex process with multiple waves of admixture.
– Clustering methods are designed to find a structure in a genetic dataset but they do not necessarily reflect real shared ancestry. Further test using other methods are required to robustly support one hypothesis.
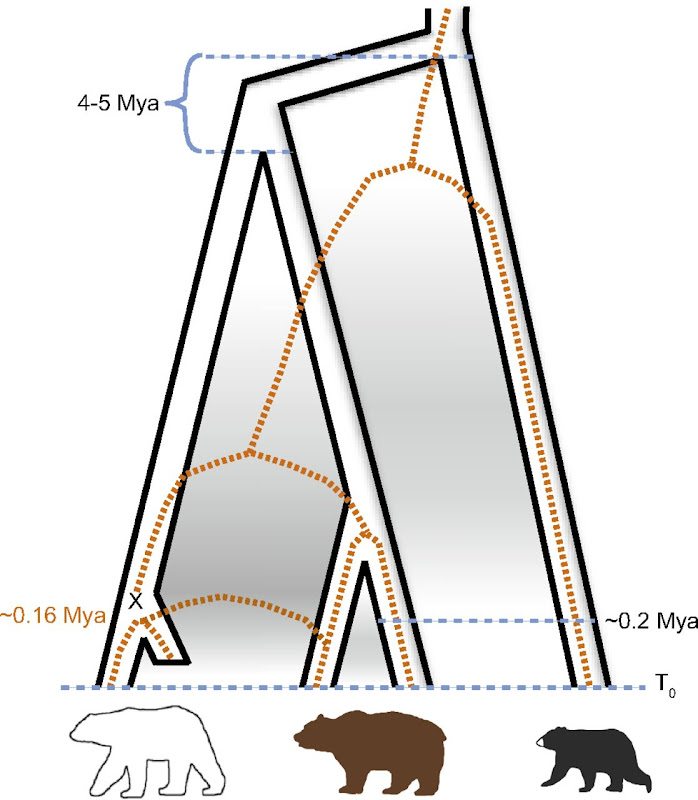
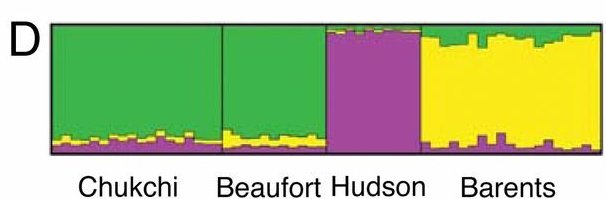
]]>- Polar bear underwent a very rapid and recent (less than 200 ky ago) adaptation to extreme environment (previously not seen in mammals)
- Brown bear is a paraphyletic taxon, as polar bear is the sister specie of the ABC bears (see Fig. 1)
 |
||
| Fig. 1: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
| Fig. 2: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
 |
||
| Fig. 3: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
 |
|
| Fig. 4: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
 |
| Fig. 5: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
 |
||
| Fig. 5: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
 |
| Fig. 6: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
I think that to address the subject of adaptation in polar bear, a study of positive selection in protein-coding gene is lacking. As authors already conducted transcriptome sequencing of polar and brown bears, annotating gene in their genome, selecting orthologous genes together with other copies from completely sequenced genomes, as dog, panda and other mammals, and then using a model to test for positive selection such as implemented in PAML would be an efficient way to identify genes of interest in the polar (or ABC) bears. Nevertheless, I am very well aware of the tremendous amount of work already performed in this PNAS paper.
 |
| Fig. 6: Miller et al., Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change, PNAS 2012 |
- The bump in the polar bear curve signified as the “Post Eemian increase” was not significant when looking at the 95% interval range in the supplementary material
- Knowing from the previous part of the article the extended hybridization between ABC and polar bears, would not the diversity introduced during those event affect the effective population size reconstruction ?
Hailer F, Kutschera VE, Hallström BM, Klassert D, Fain SR, Leonard JA, Arnason U, & Janke A (2012). Nuclear genomic sequences reveal that polar bears are an old and distinct bear lineage. Science (New York, N.Y.), 336 (6079), 344-347 PMID: 22517859
Miller W, Schuster SC, Welch AJ, Ratan A, Bedoya-Reina OC, Zhao F, Kim HL, Burhans RC, Drautz DI, Wittekindt NE, Tomsho LP, Ibarra-Laclette E, Herrera-Estrella L, Peacock E, Farley S, Sage GK, Rode K, Obbard M, Montiel R, Bachmann L, Ingólfsson O, Aars J, Mailund T, Wiig O, Talbot SL, & Lindqvist C (2012). Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proceedings of the National Academy of Sciences of the United States of America, 109 (36) PMID: 22826254