
![]() With the rise of genomics and the availability of whole genome sequences, geneticists hope to be able to understand the recent adaptations humans underwent. Classic selective sweeps, where a beneficial allele arises in a population and subsequently goes to fixation, leave a specific pattern. Indeed, all variation is erased as the selected allele invades the population, and the neighboring neutral variation is also partially swept, with an intensity depending on the linkage with the selected region.
With the rise of genomics and the availability of whole genome sequences, geneticists hope to be able to understand the recent adaptations humans underwent. Classic selective sweeps, where a beneficial allele arises in a population and subsequently goes to fixation, leave a specific pattern. Indeed, all variation is erased as the selected allele invades the population, and the neighboring neutral variation is also partially swept, with an intensity depending on the linkage with the selected region.
 |
| An example of classic selective sweep pattern. As the distance from the selected nucleotide increases, diversity increases. Fig. 2 from Hernandez et al. 2011. |
The selective sweep pattern was used to find evidence for recent adaptation in humans. Many candidate genes for recent adaptation in humans were found. Nevertheless, the preeminence of classic selective sweeps compared with other modes of adaptation (like background selection or recurrent a.k.a. “soft” sweeps) is still unknown.
In this paper, the authors claim that classic selective sweeps are in fact a rare event in human recent evolution. They argue that the overall pattern found in genome scan studies can be explained with only nearly neutral mechanisms (neutral evolution plus some purifying selection), without any positive selection going on. This casts a doubt on our ability to detect regions under selection from molecular data with currently available techniques.
Their evidence is based on polymorphism data from 179 human genomes from the 1000 genome project (see Durbin et al. 2010). The authors identified single nucleotide polymorphism. They pooled together all exons in order to see the overall sweep pattern around each substitution. The first blow to the preeminence of classic selective sweeps comes from the fact that synonymous and non-synonymous sites show the exact same sweep pattern. We would expect that non-synonymous sites, as they should be the targets of adaptation, show a stronger sweep pattern. Another concern comes from the comparison of genetic data with the expectation under neutral evolution. They show (see fig. 3) that if classic selective sweeps are frequent (more than 10% of human specific substitutions), we have the statistical power to detect a difference with a purely neutral evolution scenario. Nevertheless, we do not observe any difference between the genomic data and the neutral simulations.
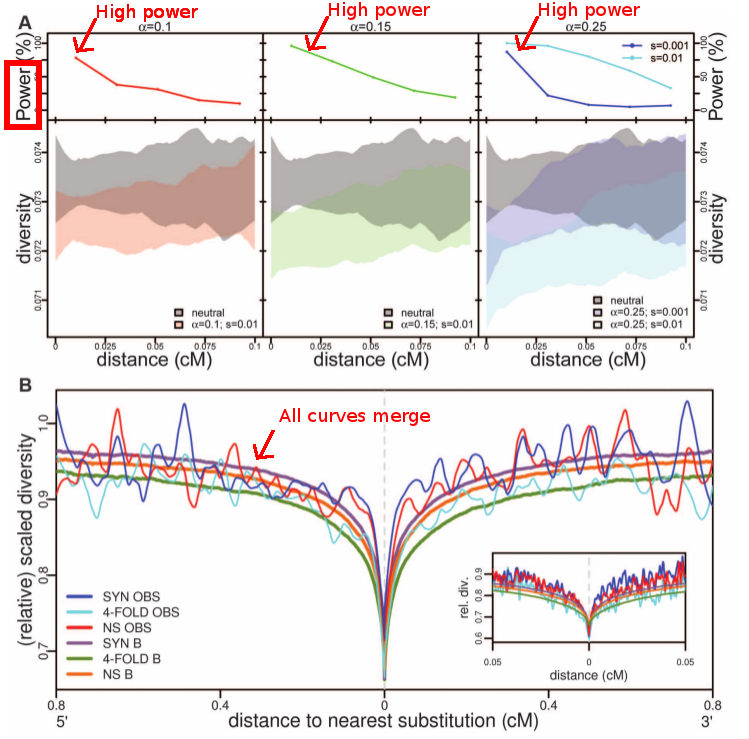 |
| Comparison of simulations under a neutral model with a model with selection, and the actual human genomes data. What is interesting in panel A is that the power is strong for all fractions of the genome under selection the authors tested (alpha parameter). Therefore the authors claim that if classic selective sweeps are frequent in the population, we should be able to detect a significant departure from neutrality. Panel B completes the argument as we can see that all curves (neutral model and human genome data) are merged. Considering that we should have the power to detect a departure from neutrality, the authors claim that the neutral scenario cannot be rejected. Fig. 3 from Hernandez et al. 2011. |
They conclude that classic selective sweeps should not have been the major mode of adaptation in recent human evolution.
I personally was not convinced by the relevance of using a mean pattern, over all coding regions, to attest that classic sweeps were rare in human evolution. Indeed, most coding regions have not experienced a selective sweep in the past, and thus the mean pattern should indeed not differ from a neutral or background selection model. Nevertheless, the authors anticipated this argument, as they run simulations where only a fraction of the genome is under positive selection. And as I wrote above, they show that we should be able to discriminate between selection and background mutation, even if the proportion of loci under selection are as low as 10% of human specific substitutions.
We raised during our discussion another concern, regarding the parameter range covered in their simulations. Indeed, the authors tested the power to distinguish selection and neutrality with several fractions of the genome under positive selection, but did not test a wide range of selection coefficient. A selection coefficient of 0.01 already seems very large, and the question remains to see if with weaker selection, we do expect to see a difference in the mean pattern of diversity over all exon SNPs.
In conclusion, I believe that the authors showed that so far we can only detect classic AND very strong selective sweeps from molecular data. In my opinion, this means that we can rarely detect classic selective sweeps. The question remains whether classic but weaker selective sweeps were rare in recent human evolution.
Hernandez, R., Kelley, J., Elyashiv, E., Melton, S., Auton, A., McVean, G., , ., Sella, G., & Przeworski, M. (2011). Classic Selective Sweeps Were Rare in Recent Human Evolution Science, 331 (6019), 920-924 DOI: 10.1126/science.1198878