
![]()

“ Hunting and gathering was humanity’s first and most successful adaptation, occupying at least 90 percent of human history. Until 12,000 years ago, all humans lived this way. ”
[The Cambridge Encyclopedia of Hunters and Gatherers. Richard B. Lee and Richard Daly, 1999]
Despite of playing a central role in human evolution, African populations remain one of the most understudied groups in human genomics. Furthermore, African human populations preserve the most genetic diversity in the world, and the study of this genetic diversity among the multitude of diverse African ethnic groups is crucial for reconstructing modern human origins. The succession of African hunter-gatherer societies is the longest and one of the most varied known, and African hunter-gatherer populations have some of the deepest divergence time of our species.
In this work the genomes of 15 African hunter-gatherers were sequenced at high coverage, expanding the catalog of human genetic variation and increasing the few number of high coverage African genomes that have been analyzed to date.
Samples and population features
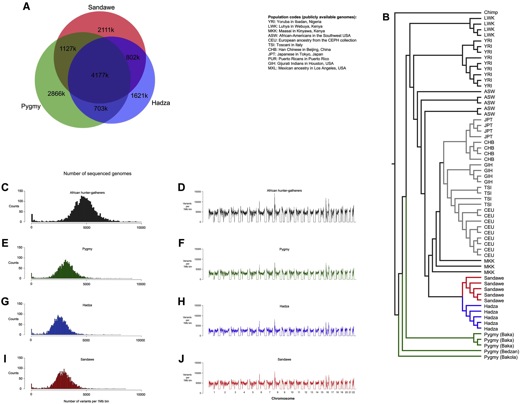
The 15 sequenced genomes were of male individuals coming from three different hunter-gatherer populations: 5 Pygmies from Cameroon, 5 Hadza and 5 Sandawe from Tanzania (Fig. 1). The Hadza and Sandawe are geographically close and they both speak the khoesan language; even if classified with the same name, their languages are highly different from each other and from khoesan languages spoken by Southern African San populations. The forest-dwelling, short-statured Pygmies are separated from the other two populations; their ancestral language has been lost after the expansion of Bantu-speaking agriculturalists within the last few thousand years, and today they are highly admixed with the Bantu. Due to Bantu’s expansion, many Sandawe, that count today more than 30000 individuals, have switched from nomadic hunting to agricultural of subsistence; on the contrary, the majority of Hadza are still hunter-gatherers, and they currently number 1000 individuals. The Hadza, Sandawe, Pygmy and San populations have ancient divergence, but they also share lineages that are rare in other populations.
Whole-genome sequencing and compared data
Hunter-gatherer genomes were sequenced at >60x high coverage, which means that each position in the human genome was sequenced 60 times, on average; two additional test of genotyping accuracy revealed low error rates, and quality control filters were applied to the data. These 15 African genomes were compared to a published genome sequence from a San hunter-gatherer and to publicly available genomes from 69 individuals, 18 from diverse African population and 51 from non-Africans (the composition of the samples is available here http://www.completegenomics.com/public-data/69-Genomes/ and more specifically in the pdf available on the page).

Genetic diversity of African hunter-gatherers
From the sequenced genomes the researchers identified 13.4 million variants (variants are the SNPs, insertions and deletions that differ from the reference sequence of the human genome GRCh37/hg19). Of these variants, 3 million were not present in the 1000 Genomes Project release of October 2011, thus the work provide the expansion of the catalog of genetic variation.
The three hunter-gatherer populations share the 50.8% of variants; it results also that genomic regions with high content of variants in one population contain a large number of variants in other populations (Fig. 2: A, C-J). Among the hunter-gatherer populations, Pygmies contain the most genetic variation, followed by the Sandawe and the Hadza. A significant genetic diversity is maintained between the populations, also between the Hadza and Sandawe that are geographically close (~150 Km) and both Khoesan speakers. The Hadza and Sandawe in fact do not share an excess of variants, and this is consistent with an ancient divergence and different mtDNA haplogroups.
Shared ancestry and demographic variation
To assess shared ancestry between different hunter-gatherer populations, the San genome was taken into account. The amount of shared variants between the populations suggests that the San diverged before the other populations, and from a test of admixture it is possible to deduce that the Hadza and Sandawe may have diverged more recently in the past than the Pygmy/San split. This is in line with the neighbor joining tree, that shows Pygmies diverging before the Hadza and Sandawe split. Pygmies are basal to the other populations, and show also population substructure (Fig. 2B).
The neighbor joining tree is complement to the principal component analysis, that was run on the 15 hunter-gatherer genomes and 53 high-coverage genomes from the Complete Genomics public data (a subset of the previous 69 individuals). The first principal component analyses distinguishes Africans from non-Africans, and the second divides Asian from European; with PC3 Hadza are differentiated from other African populations, and the following PCA differentiates Pygmies and Sandawe from the other African populations.
Moreover, for each hunter-gatherer genome the number and cumulative size of long runs of homozigosity (ROH) was calculated. The five genomes with most ROH all belong to Hadza population, and an explanation to this pattern is a population bottleneck, or an additional cause is inbreeding among the Hadza. The result from PC3, that differentiate the Hadza from other Africans populations, may also be explained by population bottleneck and/or inbreeding in the Hadza, and consistent with this, the proportion of polymorphic sites is lowest for the Hadza (and highest for Pygmies).
Local adaptation
To check if the three hunter-gatherer populations show local adaptation, the authors compared the genomes of each population with the genomes of agricultural Yoruba and pastoral Maasai populations. Using locus-specific branch lengths (LSBL), they identified 100kb windows that are highly divergent between the African hunter-gatherer populations and the Yoruba and Maasai populations. The LSBL at each polymorphic site is based on FST, and large LSBL occur when the allele frequencies of hunter-gatherer populations are very different form agricultural and pastoral populations.
What emerged from this analysis is that hunter-gatherer populations show local adaptation, with high LSBL for genes involved in smell and taste, pituitary development, reproduction and immunity. This suggests that populations are adapted to different resources, pathogens and environments.
Population-specific adaptation and population demographic factors can be evaluated by the AIMs (ancestry informative markers), population-specific variants that are at high frequency in genomes. In the hunter-gatherers’ genomes the AIMs are not randomly distributed, and they are highest in the Hadza, which is consistent with a population bottleneck and greater isolation than the other hunter-gatherer populations.
In the Pygmies, LSBL and AIM cluster analyses were used to identify variants involved in short stature, a trait that is supposed to be an adaptation to a tropical forest environment. The researchers found the largest Pygmy AIM cluster on the third chromosome, and within this cluster Jarvis et al. (2012) had previously found a 15 Mb region that shows high level of differentiation between Pygmies and the neighbor Bantu populations and is associated with height in Pygmies. Moreover, one of the genes lying within this cluster is involved in the development of the anterior pituitary, the site of synthesis and secretion of the growth hormone. Pygmies are ~17 cm shorter on average than their Bantu neighbors and among the shortest populations globally, to give an idea Pygmy men stand just 150 cm on average (Jarvis et al., 2012). Several theories have been proposed based on natural selection to explain Pygmies’ short stature, as thermoregulation, lowered nutritional requirements, or easier mobility in a dense forest environment.
Archaic admixture
The authors detected putatively introgressed region in the three hunter-gatherer populations. To do that, they modified a statistic previously used to detect archaic admixture, and tested its robustness. They found evidence of an introgression event predating the divergence of the three hunter-gatherer populations, as low levels of introgression from an unknown archaic population or populations occurred in the examined samples.
To conclude, with this work the researchers found that the three African hunter-gatherer genomes contain more variants in comparison with non-African ones, and that Pygmies contain the most genetic variation. They also observe local adaptation in the three populations, which reflect different local selective pressure. This means that Hadza, Sandawe and Pygmy populations have continued to adapt to local conditions while maintaining their own cultures of hunting and gathering.
They also found evidence that actual hunter-gatherer genomes contain introgressed archaic sequence from at least one archaic population.
References
Jarvis et al., 2012. Patterns of ancestry, signatures of natural selection, and genetic association with stature in Western African pygmies. PLoS Genet. 8, e1002641.
Lee R. B. and Dally R., 1999. The Cambridge Encyclopedia of Hunters and Gatherers.
Lachance J, Vernot B, Elbers CC, Ferwerda B, Froment A, Bodo JM, Lema G, Fu W, Nyambo TB, Rebbeck TR, Zhang K, Akey JM, & Tishkoff SA (2012). Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell, 150 (3), 457-69 PMID: 22840920