
![]()
The polar bear (Ursus maritimus) is a carnivorous species which is closely related to the brown bear (Ursus arctos) and is adapted to the severe living conditions of the High Arctic due to the great physiological changes happened during evolutionary speciation. Despite numerous researches it is still unclear when exactly this two species diverged. That’s why, Liu with colleagues in their work tried to determine a reliable divergence time of polar bear and brown bear populations and investigated demographic history as well as selection and adaptation of polar bears.
Summary
By applying a population genomic framework the authors analyzed 89 complete nuclear genomes of polar bears and brown bears. They showed that two species diverged 479-343 thousand years ago (kya) and found 16 genes under strong positive selection on the polar bear in comparison with the brown bear. They analyzed more precisely nine of these genes that are known to be associated with high risk of cardiomyopathy and vascular diseases in humans. However, in polar bears these genes are responsible for an important reorganization of the cardiovascular system which allowed them to survive in extreme life’s conditions within Arctic Circle (e.g. very low temperatures, high physical activity in cold water, high demand of the energy, hyperlipidemic diet, etc).
Principal results and discussion
In brief, authors analyzed 79 polar bears and 10 brown bears from different areas. First of all, they sequenced and de novo assembled a polar bear reference genome. The data analysis was performed by population genomic framework.
They determined joint demographic history of polar bear and brown bear, inferred effective population sizes and estimated the divergence time of two species around 479-343 kya by using two complementary methods – identity by state (IBS) tracts of DNA and diffusion approximation for demographic interference (????) (Fig. 2, taken from original publication). Both of these approaches based on past population size changes. This principal point allows avoiding mistakes from a simple isolation-with-migration model which does not consider ancient population size changes and overestimates the divergence time. The same methods together with D statistics (the ABBA-BABA test) also allowed authors to investigate the patterns and direction of gene flow between two populations from their split.
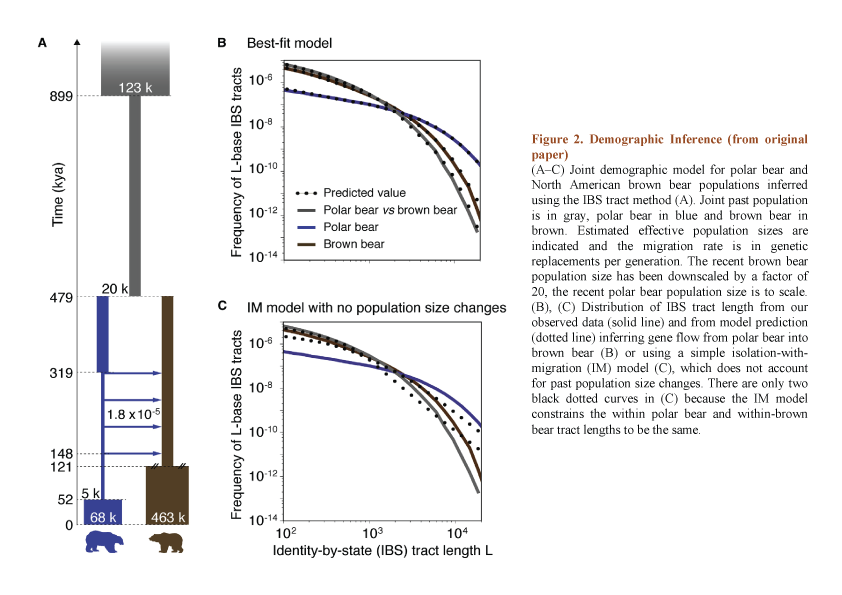
Liu and colleagues discussed evidences suggesting population bottlenecks and reduction in effective population size of polar bear that was accompanied by period of migration. Moreover, on the Figure 2a they indicate an important size reduction of joint past population predating the divergence of polar bear and brown bear. In turn, this divergence was also followed by decrease of population size in polar bear that was greater and longer than in brown bear. However, later we observe an increase of both population sizes which started earlier in brown bears.
Also, authors indicate that the gene flow corresponds to four migration waves between polar bear and brown bear populations (dated 319 – 148 kya) which probably continue till the present. However, they didn’t exclude that this migration could have occurred earlier because IBS method has some limitations to detect migration close to split time. On the Figure 2b authors constructed a best-fit model reflected that a great part of the gene flow took place from polar bear to brown bear. Apparently, these data do not correspond to results obtained with isolation-with-migration model proposed earlier (Fig.2c).
Next big part of paper is devoted to investigation of divergence and of polymorphism between polar bear and brown bear by analyzing of polar bear’s genes under positive selection that managed the reorganization of their cardiovascular system and facilitated adaptation to extreme living conditions of the High Arctic after divergence. Also, authors tried to find different evolutionary changes in protein coding sequences as well as they performed analysis of the coding regions of 19.822 genes and they used the giant panda as an outgroup. Interestingly, most of genes under positive selection were associated with vital processes such as cardiovascular function, heart development, blood coagulation, adipose tissue development and metabolism of fatty acids. However, in humans these genes are responsible for cardiomyopathy and vascular diseases.
Conclusions and personal comments
On my opinion, this paper touches a very interesting issue. Especially, I appreciated that by using genomic approaches Liu with colleagues suppose genetic causes underlying development of human cardiovascular diseases. It is an interesting point even if it needs to be proved by farther investigations, for example, by analyzing other species which are more closely related to humans.
Personally, I think that the genomic approach chosen by authors allows us to elucidate the divergence time, speciation and evolutionary history of two species of bears that were not so clear by the present time. However, from my point of view, a deeper analysis could be performed to investigate the current state of both brown bear and polar bear populations because in the paper authors indicate the possibility of recent gene flow between polar bear and brown bear.
Reference
Liu, S., Lorenzen, E., Fumagalli, M., Li, B., Harris, K., Xiong, Z., Zhou, L., Korneliussen, T., Somel, M., Babbitt, C., Wray, G., Li, J., He, W., Wang, Z., Fu, W., Xiang, X., Morgan, C., Doherty, A., O’Connell, M., McInerney, J., Born, E., Dalén, L., Dietz, R., Orlando, L., Sonne, C., Zhang, G., Nielsen, R., Willerslev, E., & Wang, J. (2014). Population Genomics Reveal Recent Speciation and Rapid Evolutionary Adaptation in Polar Bears Cell, 157 (4), 785-794 DOI: 10.1016/j.cell.2014.03.054