
![]()
Despite being the world’s most genetically diverse continent, only a handful of studies attempted to understand the genetic risks for diseases of the African populations. This study shines light not only on the genetic diversity to help learn more about the variants that are associated with malaria and hypertension, but also on the population history across sub-Saharan African populations. Beside the comprehensive map of the African variants obtained from genotypes of 1,481 individuals and whole-genome sequences of 320 individuals, authors offered a design of the array suitable to capturing variants of African populations.
Summary and comments of the paper
Population structure in SSA. Comparing ~2.2 million variants of 18 ethno-linguistic groups from sub-Saharan Africa (SSA), authors found modest differentiation among SSA populations (mean pairwise Fst = 0.019) and among Niger-Congo language groups (mean pairwise Fst = 0.009). In the article, authors suggested that the modest differentiation among Niger-Congo language group showed evidence for ‘Bantu expansion’. However, the Fig1.a shows sample distribution mostly next to the Western, East and South African coasts, rather then inside of continent where the Bantu expansion occurred, therefore indicating the sampling bias.
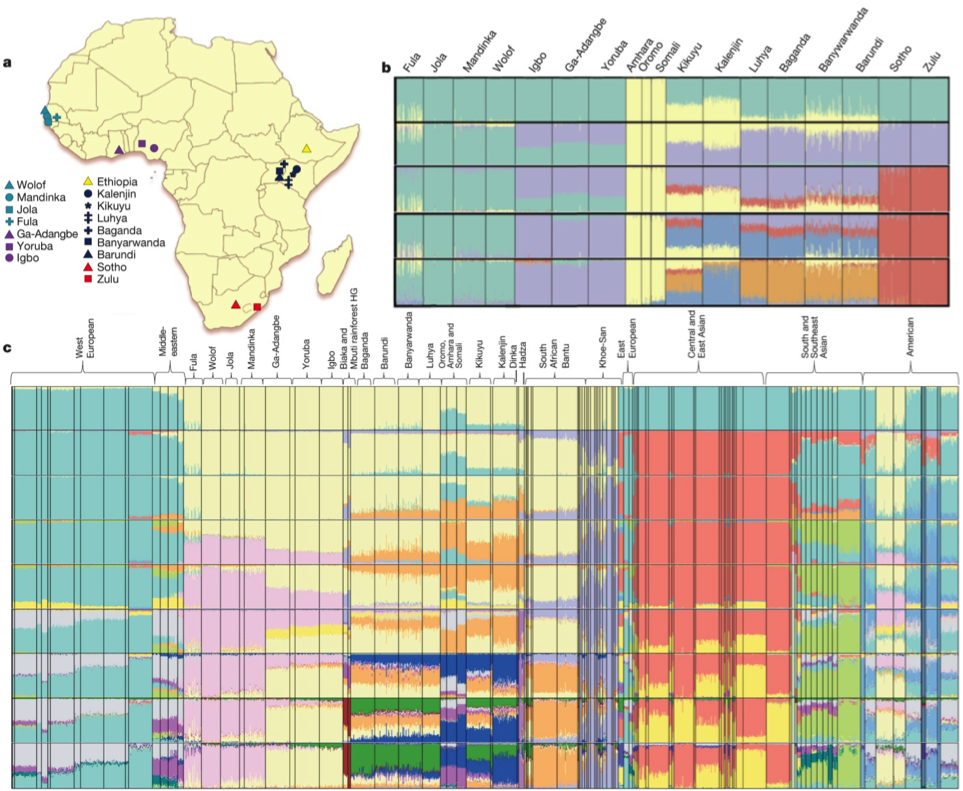
Furthermore, the authors found a high proportion of unshared and novel variants in Ethiopian population raising the importance of sequencing individuals across Africa.
Extending the analysis on population history in Africa, authors performed PCA analysis among African populations. The results suggested Euroasian gene flow and possible hunter-gatherer (HG) ancestry. To support the results from PCA analysis, unsupervised ADMIXTURE analysis (Fig1.c) showed similar results, with Euroasian admixture in Ethiopian population (Oromo, Amhara, Somali) and HG admixture in Biaka and Mbuti rainforest HG. Also, it is noticeable that Western Europeans and Central/Eastern Asian are well separated, indicating two branches of migration. ADMIXTURE analysis also pointed out the heterogeneous American population. The authors found that the most probable number of clusters of worlds populations in ADMIXTURE analysis is k = 18. Unfortunately, it is not clearly seen from the supplementary data that the CV error of clusters was lowest for k = 18.
The authors were interested in the more detailed gene flow effect among the African populations by masking Euroasian admixture. The results showed reduced population differentiation, suggesting that Euroasian admixture has a significant impact on those populations. Nevertheless, the authors did not discuss other possibilities of gene flow effects, such as allele surfing or allele fixation.
Population admixture in SSA. Using three population tests (f3 statistics), authors identified greatest proportion of Euroasian admixture in East Africa and HG admixture among Zulu and Sotho populations in South Africa. In the Fig2., authors showed that ancient Euroasian admixture appears in Yoruba population (~7,500-10,500 years), which gives support to Neanderthal ancestry in this African population.
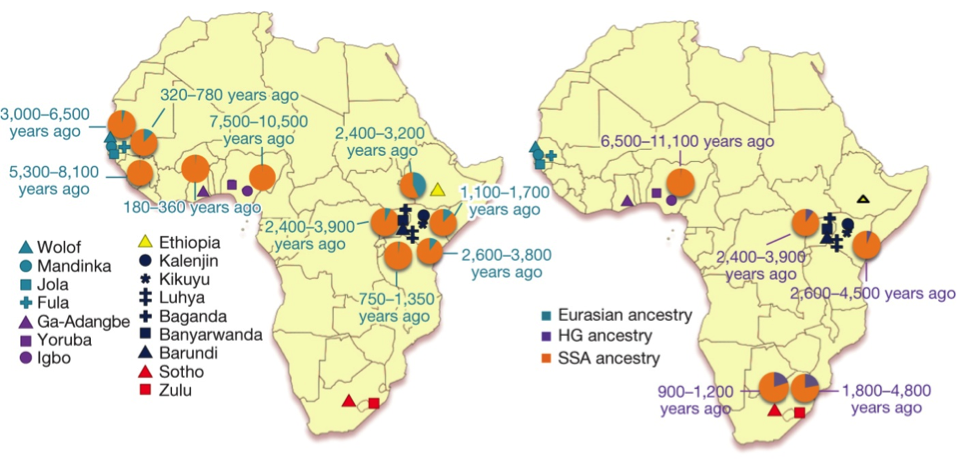
Beside the observed HG admixture in South African samples, a HG admixture was also detected in Igbo populations and more recent in East Africa. The explanation of HG admixture in West and South Africa is related to Khoe-San populations, while in the East Africa is related to Mbuti rainforest HG populations dating to ~3,000 years ago.
Moreover, in the Fig 2. is observed an overlap of Euroasian and HG admixtures in East African populations (Barundi, Banyarwanda and Baganda) both dating to ~2,400-3,900 years ago. However, it was not commented in article do these populations have a presence of both admixtures or not and how is it possible.
Positive selection in SSA. The authors observed highly differentiated SNPs in two population structure approaches to inspect the positive selection due to local adaptive forces.
One approach was to observe highly differentiated SNPs between Euroasian and African populations. Beside some other locus-specific differentiations, they found evidence of differentiation in CR1 gene (chemokine receptor 1), previously reported as a gene implicated in malaria susceptibility. The authors also identified locus-specific differentiation within genes active in osmoregulation, specifically in hypertension. Given these results, the authors speculate that changes in these gene regions give basic support in differences of salt sensitivity and hypertension in sub-Saharian African populations.
Second approach observed highly differentiated SNPs among the African populations when Euroasian admixture was masked. It has not escaped to notice that the most of Euroasian admixture had main proportion in Ethiopian populations (as seen in Fig2. and Fig1c.). For that reason, masked Euroasian admixture might affect only Ethiopian population, but certainly cannot be generalized for other African populations that actually might have had a process of local adaptation. Consequently, the quote from paper “This suggests that a large proportion of differentiation observed among African populations could be due to Euroasian admixture, rather than adaptation to selective forces.” should be taken cum grano salis. The speculative reason why there is an observed Euroasian admixture in Ethiopian population is that nomadic groups survived the migration from North and cross the Sahara to inhabit current Eastern African territory.
However, the analysis of African populations with masked Euroasian admixture revealed 56 loci, together with highly differentiated variant in CSK gene region, involved in hypertension. The variant in CKS gene region showed complete linkage disequilibrium (LD) with another risk allele that correlates with latitude, giving the evidence of temperature local adaptation as a mechanism of hypertension.
Next, the authors were interested in comparison of populations situated in endemic and non-endemic regions to distinguish loci related to infectious diseases. They identified set of loci signals in gene regions for malaria, Lassa fever, trypanosomiasis and trachoma.
Designing medical genetics studies in Africa. Taking into consideration that there is a high genetic diversity on African continent, the importance to build the reference genome panel across African populations cannot be stressed enough since it enable us to shed light on most of the worlds variation. Current reference genome panels, such as HapMap and 1000Genome, were mostly built on European, American and Asian populations and they miss the African polymorphisms. This makes more difficult to recognize certain polymorphic biomarkers associated to spectrum of diseases in African populations.
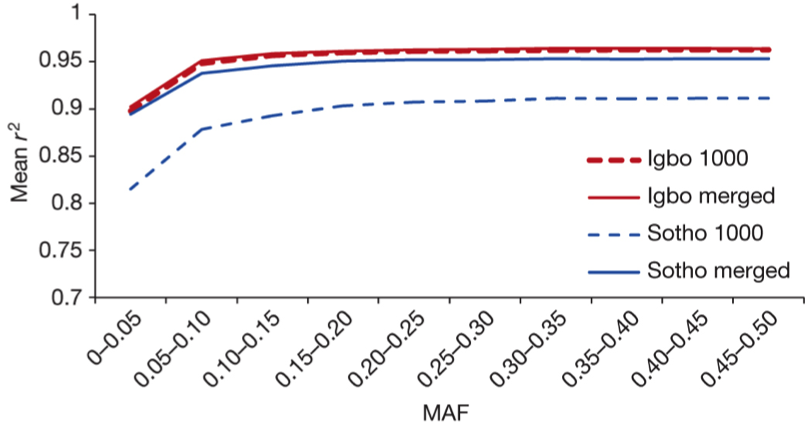
Therefore, authors investigated imputation accuracy of two African populations using two different reference genome panels – 1000Genome project and ‘merged’ 1000Genome project with 320 whole genome-sequenced African individuals, respectively. They observed the slight improvement in imputation accuracy of the Sotho and Igbo populations using ‘merged’ reference genome panel (Fig3.).
Moreover, the authors compared the usefulness of current array chips to define the most favorable array design capturing African variants. Their results showed efficiency of HumanOmni2.5M array capturing >80% of common variation. Surprisingly, authors did not mention future possibilities of whole-genome sequencing in Africa that play a crucial role in modern research nor the drawbacks of microarray noisy data. The dropping costs of sequencing technology and its development would certainly bring more precise results.
Conclusion
In spite of the nicely presented results with plenty of supplementary data, the article raises lots of speculations and thoughtful discussions on migration of African populations. Furthermore, the PCA analysis in extended and supplementary data are hard to read due to many different symbols and colors. Easier representation of PCA analysis would help to distinct the patterns of African populations. However, the study provides invaluable resource of variant association information for several diseases that will increasingly improve medical diagnostics in African populations.
Gurdasani, D., Carstensen, T., Tekola-Ayele, F., Pagani, L., Tachmazidou, I., Hatzikotoulas, K., Karthikeyan, S., Iles, L., Pollard, M., Choudhury, A., Ritchie, G., Xue, Y., Asimit, J., Nsubuga, R., Young, E., Pomilla, C., Kivinen, K., Rockett, K., Kamali, A., Doumatey, A., Asiki, G., Seeley, J., Sisay-Joof, F., Jallow, M., Tollman, S., Mekonnen, E., Ekong, R., Oljira, T., Bradman, N., Bojang, K., Ramsay, M., Adeyemo, A., Bekele, E., Motala, A., Norris, S., Pirie, F., Kaleebu, P., Kwiatkowski, D., Tyler-Smith, C., Rotimi, C., Zeggini, E., & Sandhu, M. (2014). The African Genome Variation Project shapes medical genetics in Africa Nature, 517 (7534), 327-332 DOI: 10.1038/nature13997
Please clarify this for me does it mean the Sotho and Zulu in South Africa have the same as the Igbo population in Nigeria?
Thanks
Sorry I meant does it mean that the Sotho and Zulu in South Africa have the same DNA as the Igbo population in Nigeria