
![]()
Protein abundances mainly determined by the balance of transcriptional and translational regulation. Because of the limited technology for the translational research, however, gene expression evolution was based almost entirely on studies of transcriptional regulation. With the quickly development of ribosome profiling–isolating and sequencing short fragments of mRNA bound by actively translating ribosomes–now we can study translational regulation conveniently and efficiently.
Simultaneous detection of regulatory divergence at two levels
In this paper, firstly, in order to assess the relative contributions of regulatory elements evolution to the changes in mRNA abundance and translation rate, the authors applied ribosome profiling and RNAseq to two species of Saccharomyces yeast (S. cerevisiae and S. paradoxus )and their interspecific hybrid (figure 1).
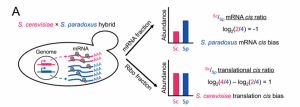
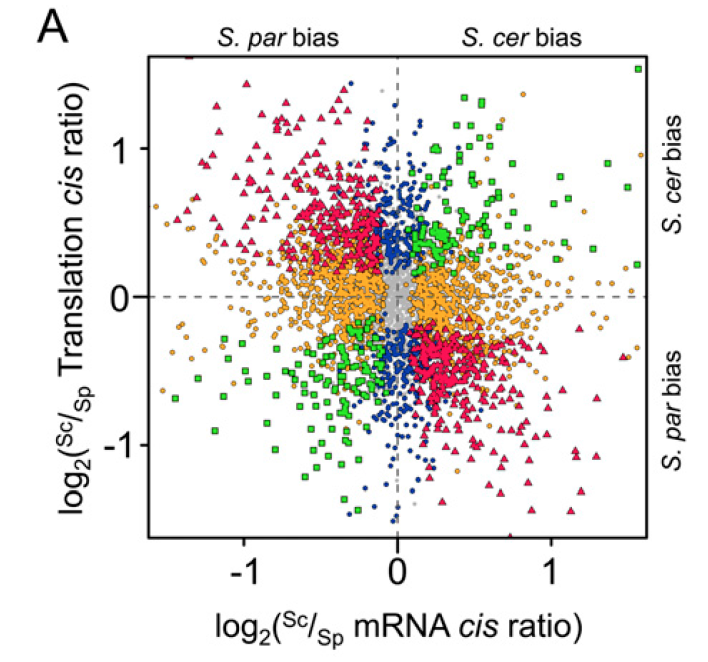
Within hybrids, both alleles share the same trans-acting cellular environment. Therefore, different mRNA abundance or translation efficiency is caused by cis-regulatory divergence. By applied these methods, the authors showed cis-regulatory divergence in both transcription and translation are abundant, almost 35% orthologs have significant divergence in translational efficiency, as compared with 61% with significant divergence in mRNA abundance. Because they identified cis-regulatory elements change at two regulatory levels simultaneously, an interesting question will be asked is whether changes at the two levels could be reinforcing (acting at the same direction) or opposing (acting in opposite directions). Compared with transcriptional divergence, surprisingly, they found the majority of translation rate divergence has an opposed effect (figure 2).In other words, it means that translational divergence acts to buffer changes in mRNA abundance, leading to maintenance of similar protein abundances between species. This phenomenon makes me quite impressive, because it shows that measuring mRNA abundance to study the expression evolution is not appropriate for some genes its protein abundance are also determined by translation rate. At the same time, the authors found that trans-acting regulatory divergence is also widespread at both regulatory levels and has an opposing pattern between the two levels.
Polygenic selection at two levels of gene regulation
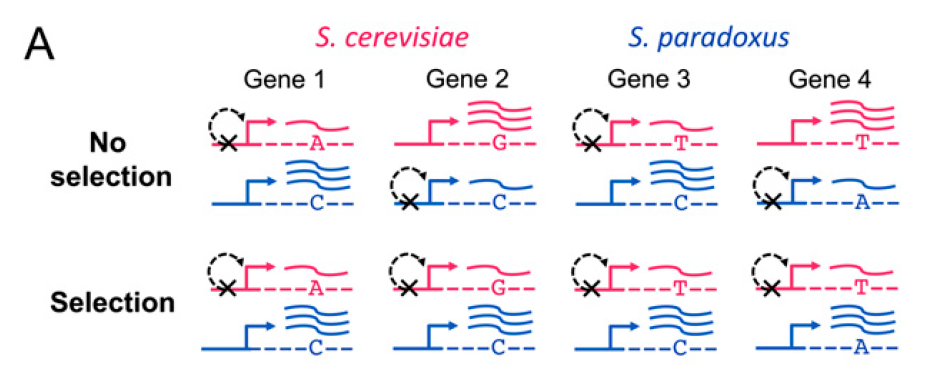
Secondly, the authors applied a recently developed approach to detect expression adaptation evolution (independently at the level of transcription and translation, as well as among all orthologs with reinforcing direction between the two regulatory levels) across functionally related groups of genes. The basic theory of this approach is based on the null hypothesis that under neutral divergence of cis-regulation, no consistent bias is expected in the relative parental direction of ASE (allele specific expression) among genes within a functional category. So, consistent directional bias across a functional group indicates that multiple independent cis-regulatory changes have altered gene expression in a coordinated fashion, and is evidence of lineage specific selection (figure 3). Totally, the authors tested 591 gene sets for deviation from neutral expected frequencies by means of a x2 test, and used a permutation framework to control for the number of tests performed. Then, the authors took S. cerevisiae strain S288c is more resistant to heavy metals than S. paradoxus strain because higher levels of both mRNA and translation in S. cerevisiae among genes whose loss leads to heavy metal sensitivity as an example to show how nature selection affected phenotype by shaping genotype. Their finding of natural selection on both levels of regulation, in some cases targeting the same gene sets, highlights the importance of considering both levels simultaneously.
Identification of conserved C-terminal peptide extensions

Liken alternative splicing, infrequent stop-codon readthrough–involves the ribosome inserting an amino acid into the growing peptide at a stop-codon position and continuing in-frame translation–is another way to increase peptide diversity. Taking advantage of multispecies riboprofiling data, next, the authors focused on searching for direct evidence of translation in putative C-terminal extensions at the transcriptome wide level. Results showed that putative C-terminal extension was detected in one or both species in 109 and 81 cases, respectively. For example, translation initiation factor eIF1A (TIF11) has conserved C-terminal extension between both species shown in (Figure 4). Tif11 is an essential protein that is involved in start codon identification whose C terminus interacts with Fun12, a GTPase also involved in initiation of translation. Stop-codon readthrough could potentially play a role in the regulation of this interaction. At the some time, they also observed several species-specific readthrough events, suggesting this may be an unappreciated source of regulatory divergence.
Personal opinions
Because the transcription variation was buffered by translation evolution, it proves this variation has some kind of harmful effect. In other words, it means the mutations caused transcription divergence between the two species are deleterious. So, I am quite wondering why these deleterious mutations got fixed among the population. In my opinion, two hypotheses may explain it.
First of all, a mutation on a trans factor may affect the gene expression of many genes. If the net fitness effect is positive, the mutation is favored by the positive selection, even though some genes’ expression may become suboptimal. Compensatory mutations provide a strategy to further improve the fitness. Alternatively, this kind of compensatory regulation was evolved to mitigate the trade-off (caused by pleiotropy) between gene expression abundance and some other traits (such as expression noise, timing, location and so on). Taking the expression abundance and noise as an example, because the regulation mechanisms of the both traits are coupled, if nature selection shapes the promoter architecture for decreasing expression noise(beneficial), it will also change the transcription abundance(deleterious). So, in order to compensate the deleterious effect caused by transcription level variation, translation rate evolved on an opposing direction.
Artieri, C., & Fraser, H. (2013). Evolution at two levels of gene expression in yeast Genome Research, 24 (3), 411-421 DOI: 10.1101/gr.165522.113