Difference between revisions of "ISA"
| (45 intermediate revisions by 4 users not shown) | |||
| Line 1: | Line 1: | ||
| − | [[ISA tutorial]] | + | [[Category:Bulletins]] |
| − | [[Running ISA in parallel]] | + | <newstitle>ISA application note</newstitle> |
| + | <teaser>A <a href="http://bioinformatics.oxfordjournals.org/cgi/content/abstract/btq130">new application note</a> has been published recently in Bioinformatics, about the '''isa''' and '''eisa''' packages and the Iterative Signature Algorithm. | ||
| + | <date>24 Jul 2010 — 17:49</date> | ||
| + | </teaser> | ||
| + | |||
| + | [[image:expmat.png|An ISA transcription module|300px|right|link=ISA]] | ||
| + | <br/> | ||
| + | Large sets of data, like expression profile from many samples, require | ||
| + | analytic tools to reduce their complexity. | ||
| + | The '''Iterative Signature Algorithm (ISA)''' was designed to reduce the | ||
| + | complexity of very large sets of data by decomposing it into so-called | ||
| + | "modules". In the context of gene expression data these modules consist of | ||
| + | subsets of genes that exhibit a coherent expression profile only over a | ||
| + | subset of microarray experiments. Genes and arrays may be attributed to | ||
| + | multiple modules and the level of required coherence can be varied resulting | ||
| + | in different "resolutions" of the modular mapping. Since the ISA does not | ||
| + | rely on the computation of correlation matrices (like many other tools), it | ||
| + | is extremely fast even for very large datasets. | ||
| + | |||
| + | = Software for Gene expression data = | ||
| + | |||
| + | We developed the <code>eisa</code> [http://www.r-project.org GNU R] package to facilitate the modular analysis of gene expression data. The package uses standard [http://www.bioconductor.org BioConductor] data structures and includes various visualization tools as well. | ||
| + | |||
| + | === Requirements, download and installation === | ||
| + | |||
| + | To use <code>eisa</code> you will need a working [http://www.r-project.org GNU R] installation. | ||
| + | |||
| + | As of the 23rd of April, 2010, the <code>eisa</code> package is an official [http://www.bioconductor.org BioConductor] package. | ||
| + | |||
| + | <code>eisa</code> depends on a number of other R packages: <code>isa2</code>, <code>Biobase</code>, <code>AnnotationDbi</code>, <code>Category</code>, <code>genefilter</code>, <code>DBI</code>. The good news is that all these dependencies are installed automatically, and all you need to do is to start R and type in | ||
| + | |||
| + | source("http://bioconductor.org/biocLite.R") | ||
| + | biocLite("eisa") | ||
| + | |||
| + | at your R prompt. See [http://bioconductor.org/packages/release/bioc/html/eisa.html the eisa package page at the BioConductor website] for details. | ||
| + | |||
| + | Alternatively, you can also download the package from here: | ||
| + | |||
| + | * '''[http://www.unil.ch/cbg/homepage/downloads/eisa_1.0.0.zip Microsoft Windows (32 bit)]''' <br/>Download [http://www.unil.ch/cbg/homepage/downloads/eisa_1.0.0.zip this file], save it in a temporary directory, and then start R. From the Packages menu choose '<code>Install packages from local zip files</code>' and select the saved file. | ||
| + | * '''[http://www.unil.ch/cbg/homepage/downloads/win64/eisa_1.0.0.zip Microsoft Windows (64 bit)]''' <br/>Download [http://www.unil.ch/cbg/homepage/downloads/win64/eisa_1.0.0.zip this file], save it in a temporary directory, and then start R. From the Packages menu choose '<code>Install packages from local zip files</code>' and select the saved file. | ||
| + | * '''[http://www.unil.ch/cbg/homepage/downloads/eisa_1.0.0.tgz Mac OSX (Leopard)]''' <br/> Download and install [http://www.unil.ch/cbg/homepage/downloads/eisa_1.0.0.tgz this file]. | ||
| + | * '''[http://www.unil.ch/cbg/homepage/downloads/eisa_1.0.0.tar.gz Linux and Unix systems, R source package]''' <br/> Download [http://www.unil.ch/cbg/homepage/downloads/eisa_1.0.0.tar.gz this file], save it in a temporary directory, and start R. Install the downloaded package using the <code>install.packages()</code> function: give the full path of the saved file and use the '<code>repos=NULL</code>' argument of <code>install.packages()</code>. | ||
| + | |||
| + | === License === | ||
| + | |||
| + | The eisa package is licensed under the GNU General Public License, version 2 or later. For details, see http://www.gnu.org/licenses/old-licenses/gpl-2.0.html. | ||
| + | |||
| + | = Software for any tabular data = | ||
| + | |||
| + | The ISA can be applied to identify coherent substructures (i.e. modules) from any rectangular matrix of data. You can use the <code>isa2</code> R package for such an analysis. | ||
| + | |||
| + | === Requirements === | ||
| + | |||
| + | No additional R package is required to install and use <code>isa2</code>. But on Linux and Unix systems you will need a C compiler to install it. E.g. on Ubuntu Linux you will need to install the <code>build-essential</code> package. | ||
| + | |||
| + | === Installation === | ||
| + | |||
| + | The <code>isa2</code> package is available from [http://cran.r-project.org/ CRAN], the standard R package repository. You can install it on any platform that is supported by GNU R, e.g. Microsoft Windows, Mac OSX and Linux systems. To install it, start R and type in | ||
| + | |||
| + | install.packages("isa2") | ||
| + | |||
| + | at the prompt. On Linux and Unix-like systems, you will need a working C compiler for a successful installation. | ||
| + | |||
| + | === License === | ||
| + | |||
| + | The <code>isa2</code> package is licensed under the Creative Commons Attribution-Noncommercial-Share Alike 3.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/ or send a letter to Creative Commons, 171 Second Street, Suite 300, San Francisco, California, 94105, USA. | ||
| + | |||
| + | = Tutorials = | ||
| + | |||
| + | ===[[EISA tutorial|The Iterative Signature Algorithm for Gene Expression Data]]=== | ||
| + | Shows the typical steps of modular analysis, from loading you expression data to the visualization of transcription modules.<br/> | ||
| + | [[EISA tutorial|HTML]] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/EISA_tutorial.pdf PDF] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/EISA_tutorial.Rnw Rnw] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/EISA_tutorial.R R code] | ||
| + | |||
| + | ===[[EISA and the biclust package|ISA and the biclust package]]=== | ||
| + | The <code>biclust</code> package implements several biclustering algorithms. It is possible to convert the results of <code>biclust</code> to transcription modules and vice-versa. <br/> | ||
| + | [[EISA and the biclust package|HTML]] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/EISA_biclust.pdf PDF] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/EISA_biclust.Rnw Rnw] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/EISA_biclust.R R code] | ||
| + | |||
| + | ===[[Tissue specific expression with the Iterative Signature Algorithm]]=== | ||
| + | [[Tissue specific expression with the Iterative Signature Algorithm|HTML]] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/tissues.pdf PDF] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/tissues.Rnw Rnw] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/tissues.R R code] | ||
| + | |||
| + | ===[[EISA module trees|Hierarchical module trees]]=== | ||
| + | A module tree is the hierarchical modular organization of a data set.<br/> | ||
| + | [[EISA module trees|HTML]] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/EISA_module_trees.pdf PDF] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/EISA_module_trees.Rnw Rnw] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/EISA_module_trees.R R code] | ||
| + | |||
| + | ===[[ISA tutorial|The Iterative Signature Algorithm]]=== | ||
| + | Tutorial for the analysis of tabular data with the <code>isa2</code> R package. <br/> | ||
| + | [[ISA tutorial|HTML]] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/ISA_tutorial.pdf PDF] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/ISA_tutorial.Rnw Rnw] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/ISA_tutorial.R R code] | ||
| + | |||
| + | ===[[Running ISA in parallel]]=== | ||
| + | Shows how to run ISA on a computer cluster or multi-processor machine, using MPI and the <code>Rmpi</code> and <code>snow</code> R packages. <br/> | ||
| + | [[Running ISA in parallel|HTML]] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/ISA_parallel.pdf PDF] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/ISA_parallel.Rnw Rnw] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/ISA_parallel.R R code] | ||
| + | |||
| + | ===[[ISA internals]]=== | ||
| + | [[ISA internals|HTML]] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/ISA_internals.pdf PDF] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/ISA_internals.Rnw Rnw] | ||
| + | [http://www2.unil.ch/cbg/homepage/downloads/ISA_internals.R R code] | ||
| + | |||
| + | =Matlab package= | ||
| + | You can download it from [[Media:ISApackage-1.03.zip|here]]. It also includes the implementation of the Ping-pong algorithm <cite>Kutalik2008NB</cite>. The "testPP.m" file explains how the algorithm is applied to a pair of toy data sets. To test the ISA functionalities, the "testISA.m" needs to be launched. | ||
| + | |||
| + | = Papers = | ||
| + | <pubmed> | ||
| + | 18464786 | ||
| + | 15606968 | ||
| + | 15044247 | ||
| + | 14737187 | ||
| + | 12689096 | ||
| + | 12134151 | ||
| + | </pubmed> | ||
| + | |||
| + | ''PDF files:" [[Media:PPA.pdf|Kutalik2008]] [[Media:review.pdf|Ihmels 2004]] [[Media:bioISA.pdf| Ihmels 2004a]] [[Media:comparative.pdf| Bergmann 2004]] [[Media:ISA.pdf|Bergmann 2003]] [[Media:SA.pdf|Ihmels2002]] | ||
Latest revision as of 10:38, 1 February 2016
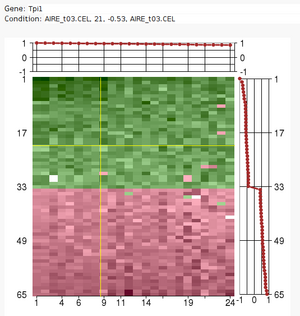
Large sets of data, like expression profile from many samples, require
analytic tools to reduce their complexity.
The Iterative Signature Algorithm (ISA) was designed to reduce the
complexity of very large sets of data by decomposing it into so-called
"modules". In the context of gene expression data these modules consist of
subsets of genes that exhibit a coherent expression profile only over a
subset of microarray experiments. Genes and arrays may be attributed to
multiple modules and the level of required coherence can be varied resulting
in different "resolutions" of the modular mapping. Since the ISA does not
rely on the computation of correlation matrices (like many other tools), it
is extremely fast even for very large datasets.
Contents
Software for Gene expression data
We developed the eisa GNU R package to facilitate the modular analysis of gene expression data. The package uses standard BioConductor data structures and includes various visualization tools as well.
Requirements, download and installation
To use eisa you will need a working GNU R installation.
As of the 23rd of April, 2010, the eisa package is an official BioConductor package.
eisa depends on a number of other R packages: isa2, Biobase, AnnotationDbi, Category, genefilter, DBI. The good news is that all these dependencies are installed automatically, and all you need to do is to start R and type in
source("http://bioconductor.org/biocLite.R")
biocLite("eisa")
at your R prompt. See the eisa package page at the BioConductor website for details.
Alternatively, you can also download the package from here:
- Microsoft Windows (32 bit)
Download this file, save it in a temporary directory, and then start R. From the Packages menu choose 'Install packages from local zip files' and select the saved file. - Microsoft Windows (64 bit)
Download this file, save it in a temporary directory, and then start R. From the Packages menu choose 'Install packages from local zip files' and select the saved file. - Mac OSX (Leopard)
Download and install this file. - Linux and Unix systems, R source package
Download this file, save it in a temporary directory, and start R. Install the downloaded package using theinstall.packages()function: give the full path of the saved file and use the 'repos=NULL' argument ofinstall.packages().
License
The eisa package is licensed under the GNU General Public License, version 2 or later. For details, see http://www.gnu.org/licenses/old-licenses/gpl-2.0.html.
Software for any tabular data
The ISA can be applied to identify coherent substructures (i.e. modules) from any rectangular matrix of data. You can use the isa2 R package for such an analysis.
Requirements
No additional R package is required to install and use isa2. But on Linux and Unix systems you will need a C compiler to install it. E.g. on Ubuntu Linux you will need to install the build-essential package.
Installation
The isa2 package is available from CRAN, the standard R package repository. You can install it on any platform that is supported by GNU R, e.g. Microsoft Windows, Mac OSX and Linux systems. To install it, start R and type in
install.packages("isa2")
at the prompt. On Linux and Unix-like systems, you will need a working C compiler for a successful installation.
License
The isa2 package is licensed under the Creative Commons Attribution-Noncommercial-Share Alike 3.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/ or send a letter to Creative Commons, 171 Second Street, Suite 300, San Francisco, California, 94105, USA.
Tutorials
The Iterative Signature Algorithm for Gene Expression Data
Shows the typical steps of modular analysis, from loading you expression data to the visualization of transcription modules.
HTML
PDF
Rnw
R code
ISA and the biclust package
The biclust package implements several biclustering algorithms. It is possible to convert the results of biclust to transcription modules and vice-versa.
HTML
PDF
Rnw
R code
Tissue specific expression with the Iterative Signature Algorithm
Hierarchical module trees
A module tree is the hierarchical modular organization of a data set.
HTML
PDF
Rnw
R code
The Iterative Signature Algorithm
Tutorial for the analysis of tabular data with the isa2 R package.
HTML
PDF
Rnw
R code
Running ISA in parallel
Shows how to run ISA on a computer cluster or multi-processor machine, using MPI and the Rmpi and snow R packages.
HTML
PDF
Rnw
R code
ISA internals
Matlab package
You can download it from here. It also includes the implementation of the Ping-pong algorithm Kutalik2008NB. The "testPP.m" file explains how the algorithm is applied to a pair of toy data sets. To test the ISA functionalities, the "testISA.m" needs to be launched.
Papers
Kutalik Z, Beckmann JS, Bergmann S
A modular approach for integrative analysis of large-scale gene-expression and drug-response data.
Nat Biotechnol: 2008 May, 26(5);531-9
[PubMed:18464786]
[WorldCat.org:
ISSN
ESSN
]
[DOI]
( p)
Ihmels JH, Bergmann S
Challenges and prospects in the analysis of large-scale gene expression data.
Brief Bioinform: 2004 Dec, 5(4);313-27
[PubMed:15606968]
[WorldCat.org:
ISSN
ESSN
]
[DOI]
( p)
Ihmels J, Bergmann S, Barkai N
Defining transcription modules using large-scale gene expression data.
Bioinformatics: 2004 Sep 1, 20(13);1993-2003
[PubMed:15044247]
[WorldCat.org:
ISSN
ESSN
]
[DOI]
( o)
Bergmann S, Ihmels J, Barkai N
Similarities and differences in genome-wide expression data of six organisms.
PLoS Biol: 2004 Jan, 2(1);E9
[PubMed:14737187]
[WorldCat.org:
ISSN
ESSN
]
[DOI]
( o)
Bergmann S, Ihmels J, Barkai N
Iterative signature algorithm for the analysis of large-scale gene expression data.
Phys Rev E Stat Nonlin Soft Matter Phys: 2003 Mar, 67(3 Pt 1);031902
[PubMed:12689096]
[WorldCat.org:
ISSN
ESSN
]
[DOI]
( o)
Ihmels J, Friedlander G, Bergmann S, Sarig O, Ziv Y, Barkai N
Revealing modular organization in the yeast transcriptional network.
Nat Genet: 2002 Aug, 31(4);370-7
[PubMed:12134151]
[WorldCat.org:
ISSN
ESSN
]
[DOI]
( o)
PDF files:" Kutalik2008 Ihmels 2004 Ihmels 2004a Bergmann 2004 Bergmann 2003 Ihmels2002