 Microbial species are one of the most ubiquitous living group on Earth’s biosphere, showing incredible ability to thrive even in ambient conditions to the limit of human endurance. By virtue of their rapid growth, bacteria are ideal for unraveling the molecular mechanisms of many evolutionary processes. Their rapidity to respond to changes has been associated to the combined effect of evolutionary processes, species composition or gene expression shifts. Most of the studies have focused so far on isolation and comparison of cultured bacterial population, while very few data are available concerning free-living bacteria. Therefore, it is still controversial how quickly, to which extent and by which mechanism microorganisms evolve in their natural environment.
Microbial species are one of the most ubiquitous living group on Earth’s biosphere, showing incredible ability to thrive even in ambient conditions to the limit of human endurance. By virtue of their rapid growth, bacteria are ideal for unraveling the molecular mechanisms of many evolutionary processes. Their rapidity to respond to changes has been associated to the combined effect of evolutionary processes, species composition or gene expression shifts. Most of the studies have focused so far on isolation and comparison of cultured bacterial population, while very few data are available concerning free-living bacteria. Therefore, it is still controversial how quickly, to which extent and by which mechanism microorganisms evolve in their natural environment.
Two researchers of the University of California, Denef and Banfield, have tried to answer this question, as described in a paper recently published in Science. Their work report evolutionary rate estimates from bacterial populations living in a really challenging site, the hot, humid, low-pH, metal-rich and low-oxygen acid mine drainage in the Richmond Mine (Iron Mountain, CA), over the course of 9 years. This would seem not the ideal model system for conducting such kind of study, for the low accessibility at the sites only in limited periods of the year, but it perfectly fits the requirement of a discrete, reproducible and simple microbial community meeting very restricted input from other regions. In fact, the air-solution interface biofilm community consists in few organisms types, normally four to six. In this case Leptospirillum group II, which comprises iron-oxidizing bacteria that can live in sulfuric acid, dominates. The authors tried to trace back a lineage history of the group, starting by using the data of previous metagenomic studies Simmons et al. from the same lab published on Plos Biology in 2008. These led to the reconstruction of Leptospirillum group II type I and VI “reference” genomes, which share about 94% average nucleotide identity. As shown in figure 1, the two populations were sampled in 2002 and 2005 at 5-way and UBA locations, respectively.
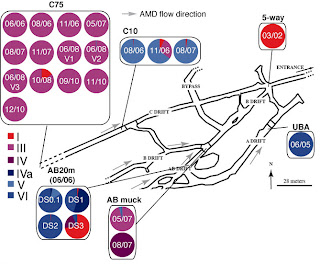
Figure 1. Adapted from Denef et al. Richmond Mine schematic map, with pie charts indicating genotype proportions in 24 samples, estimated on the basis of read recruitment.
Already assembled genomes were compared against total population DNA, thus allowing the authors to find other four distinct Leptospirillum genotypes (types II to V). As indicated by proteomics-based results published by the same group in 2009, some particular sites, like C75, were clearly dominated by Leptospirillum type III genotype, which appeared to be a recombinant hybrid of genotypes I and VI. The recombination points, which were found by identifying discontinuities in reads alignments with the reference genomes, were all located within genes. The population sampled at C75 was ideal to be used to calculate the substitution rate, because of low level of variation within population and across space. A high rate of substitution of 1.4 × 10?9 per nucleotide per generation was calculated, if compared to previous estimates of bacterial genome-wide short term substitution rates, which have ranged from 7.2 × 10?11 to 4.0 × 10?9. Many reasons can account for these unexpected results, which anyway match with universal mutations-per-genome size predicted by Drake; for example, they used a unique approach combining proteomics inferred genotyping dataset, population genomic time series and, for the first time, cultivation-independent population genotypes, but also they sampled a unique model system, where human and natural perturbations, combined with the low biological complexity, can both affect the evolutionary rate estimates.
Population genomic analyses suggested that the six Leptospirillum genotypes consist of a mosaic of type I and type VI genome blocks tens to hundreds of kb in length, probably recombined in a single cell and fixed in its descendants, as shown by the recurrence of the same transition points. Each genotype’s fixed mutation were used to construct the phylogenetic tree showed in figure 2, which suggests that the six Leptospirillum genotypes diverged from a common ancestor in a matter of decades (time of coalescence estimated between 2 and 44 years).
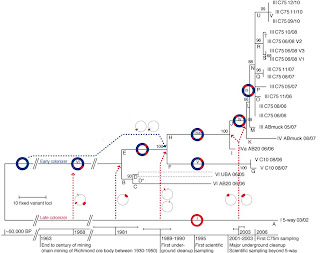
Figure 2. Adapted from Denef et al. Evolutionary history of the sampled genotypes, based on the variant loci inferred using the maximum parsimony method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test is shown next to the branches. Dotted arrows indicate recombination events; circle schematics represent the regions affected. The timeline indicates the calculated time ranges of recombination events as well as historical events. Branch D* is presented as two strains, each assigned half the total number of UBA 06/05 SNPs, because low incidence of SNPs precluded their linkage. BP, years before the present.
Nicely, evidence for positive selection for hybrid genotypes was given by the finding of fixed non-synonymous substitutions in high number of genes involved in signal transduction, transcriptional regulation and global regulators. This study suggest that the evolution of Leptospirillum consisted of a mosaic of different events, comprising homologous recombination, fixation and selective sweeps that generated the different genotypes that can be currently observed. Some limits related to the paper can be found in the final author’s speculation that states selection between genotypes as due to genotypes divergence by only a few nucleotides. In fact it’s likely that this result was biased by the approach they chose, that only relied on the comparison of genes present in their reference genomes, which can produce incomplete or erroneous interpretation if the genome assemblies are not corrected.
Denef, V., & Banfield, J. (2012). In Situ Evolutionary Rate Measurements Show Ecological Success of Recently Emerged Bacterial Hybrids Science, 336 (6080), 462-466 DOI: 10.1126/science.1218389


