 For decades, most researchers have provided some general insights into the nature of adaptation in asexually reproducing populations with small genome, such as bacteria and yeast. They assumed that sexual species evolve the same way these populations do, i.e. their adaptation is driven by the so-called selective sweeps or newly arising beneficial genetic mutation quickly becomes “fixated” on a particular portion of DNA, with the genome-wide haplotype associated with it. When we relate to obligate sexually reproducing systems, this is much more complicated by the fact that selection can act on standing variation, that means that weak selection can act on many pre-existing genetic variants involved in fitness traits. The idea is that short-term evolution have occurred through a so-called “soft sweep” model, which contrasts the hypothesis of the “hard sweep”, where strong selective sweep originates from a single mutation, while all its linked neutral variants are eliminated. Burke et al. compared outbred, sexually reproducing, replicated populations of D. melanogaster selected for accelerated development and their matched control populations on a genome-wide basis, and this is the first time that such a study of a sexually reproducing species has been done.
For decades, most researchers have provided some general insights into the nature of adaptation in asexually reproducing populations with small genome, such as bacteria and yeast. They assumed that sexual species evolve the same way these populations do, i.e. their adaptation is driven by the so-called selective sweeps or newly arising beneficial genetic mutation quickly becomes “fixated” on a particular portion of DNA, with the genome-wide haplotype associated with it. When we relate to obligate sexually reproducing systems, this is much more complicated by the fact that selection can act on standing variation, that means that weak selection can act on many pre-existing genetic variants involved in fitness traits. The idea is that short-term evolution have occurred through a so-called “soft sweep” model, which contrasts the hypothesis of the “hard sweep”, where strong selective sweep originates from a single mutation, while all its linked neutral variants are eliminated. Burke et al. compared outbred, sexually reproducing, replicated populations of D. melanogaster selected for accelerated development and their matched control populations on a genome-wide basis, and this is the first time that such a study of a sexually reproducing species has been done.
As shown in figure 1, they used the Illumina platform to get short-read sequences from three genomic DNA libraries, obtained from sets of replicated populations experiencing different selection treatments, maintained since 1980 under the specific conditions of large population size (N?>?1,000) and discrete generations:
1) a pooled sample of five replicate populations that have undergone sustained selection for accelerated development and early fertility for over 600 generations (ACO);
2) a pooled sample of five replicate ancestral control populations, which experience no direct selection on development time (CO);
3) a single ACO replicate population (ACO1).
Phenotype was assayed by using longevity assay, starvation resistance assay, development time assay, dry weight assay.
Figure 1. Grey bars represent values measured in each of the five replicate populations in the ACO and CO treatments. Measures from the five baseline (B) replicate populations represent phenotypes typical of populations kept on two-week generation maintenance schedules. Only data for females are shown. Longevity and starvation resistance data were collected after at least 619 generations of ACO treatment, and both development time and dry weight data (dry weight values are mean masses of groups of ten females) were collected after 640 generations of ACO treatment. Error bars, s.e.m. for each replicate population.
As represented in Figure 2, a 100-kb genome-wide sliding-window analysis was carried out to identify regions diverged in allele frequency, with a large number of genomic regions showing significant difference between the ACO population and their matched controls, while no significant divergence was displayed by the comparison of the single replicate population (ACO1) and the pooled sample consisting of all five ACO populations. The presence of an apparent excess of diverged regions on the X chromosome was explained as a result of selection on initially rare recessive or partially recessive alleles. Another important consideration to do is that the adaptive response was highly multigenic, as not only one or few region were identified to be affected by selection on developmental time, but most likely a larger portion of the genome was involved.
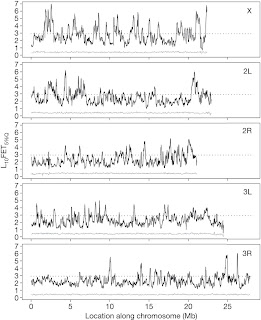
Figure 2. Sliding-window analysis (100?kb) of differentiation in allele frequency between the ACO and CO populations: the solid black line depicts L10FET5%Q scores at 2-kb steps (Methods). The dotted line is the threshold that any given window has a 0.1% chance of exceeding relative to the genome-wide level of noise. The grey line depicts L10FET5%Q scores for a difference in allele frequency between ACO1 and the ACO pooled sample. The five panels show the five major D. melanogaster chromosome arms (as indicated).
Looking instead at the heterozigosity throughout the genome, they found a relevant and expected concordance with these results. Regions of reduced heterozygosity are in fact expected to be strongly associated with regions of differentiated allele frequency. Accordingly, if we compare figure 2 with figure 3, we can observe that also in this case the regions identified for divergence in allele frequency were the ones associated with reduced heterozigosity.
Figure 3. Sliding-window analysis (100?kb) of heterozygosity in the CO pool (blue), the ACO pool (red) and ACO1(grey), with a 2-kb step size. The panels show the five major chromosome arms of D. melanogaster.
They were also able to exclude that the observed similarity in allele frequencies and patterns of heterozigosity between the ACO1 and ACO libraries was an artifact due to sample preparation or data analysis, by individually genotyping 35 females from the five replicate populations of each selection treatment at 30 loci identified for a divergence in allele frequency (Fig. 4).
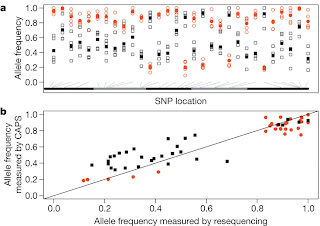
Figure 4. a, Allele frequency estimates of the most common allele at 30 SNPs genotyped in 35 females per replicate population. Red circles represent ACO estimates and grey squares represent CO estimates. Open symbols are allele frequencies for ACO1–ACO5 and CO1–CO5, and filled symbols represent treatment means. Alternating black and grey bars designate the X, 2L, 2R, 3L, and 3R arms, respectively, with grey lines indicating SNP location. b, Scatter plot comparing allele frequency estimates at the same 30 SNPs obtained from the Illumina resequencing versus individual genotyping. Red circles represent ACO, black squares represent CO and the straight line represents a slope of unity.
Burke found evidence of evolution in more than 500 genes that could be linked to a variety of traits, including size, sexual maturation and life span, indicating a gradual, widespread network of selective adaptation. There are two possible hypothesis to explain the results reported in the paper: either “parallel evolution”, with selection acting on the same intermediate-frequency variants in each population, or unwanted migration between replicate populations. In any case, these results clearly show that the signature of “classic hard sweep” is absent in this population, despite evidence of strong selection, while “soft sweep” model is more consistent with the observations.
Burke, M., Dunham, J., Shahrestani, P., Thornton, K., Rose, M., & Long, A. (2010). Genome-wide analysis of a long-term evolution experiment with Drosophila Nature, 467 (7315), 587-590 DOI: 10.1038/nature09352




