
![]()
Exploration of variability of human genomes represents a key step in the holy grail of human genetics – to link genotypes with phenotypes, it also provides insights to human evolution and history. For this purpose Exome Aggregation Consortium (ExAC) have been founded; to capture variability of human exomes using next-generation sequencing. The first ExAC dataset of 63,358 individuals was released 20th of October 2014. Recently, a paper describing updated version of the dataset was published : Analysis of protein-coding genetic variation in 60,706 humans.
Authors made a great work on the reproductibility of the downstream analyses they have performed and generally on the availability of data. All the code is well documented in blogpost and available in GitHub repository. All figures in this blogpost I plotted by my own!
Dataset
ExAC is composed of almost ten fold more individuals and previous dataset of the similar kind Fig 1a. 91,000 individuals were sequenced, of which 60,706 have been kept after quality filtering. Finnish population was excluded from European due to bottleneck they have gone though.
ExAC was targeting individuals with various genetic background. Principal component analysis have shown very strong geographical pattern in the dataset (Fig 1b). I expected a continuum of haplotypes in the environment without strong geographic obstacle (like European-Latino continuum). The gaps between South Asian samples and the rest Europen samples on the PCA plot is most likely caused by the absence of samples from Middle-East Asia. Middle-East Asian samples have just a colour, but no data points. Central Asians do not even have a colour.
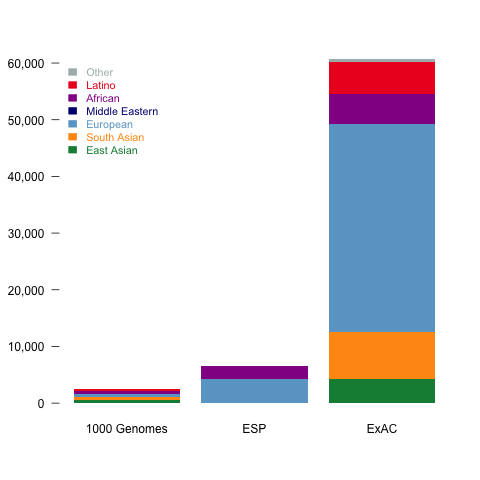
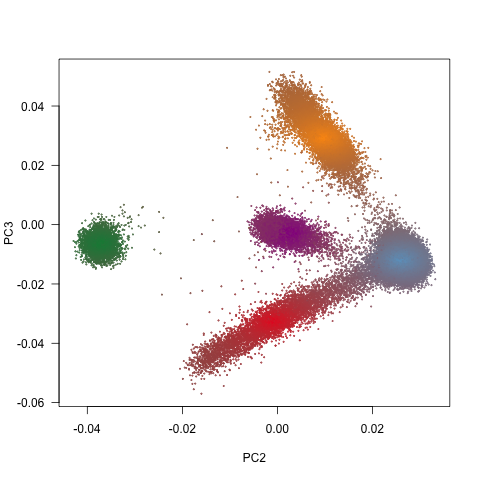
Figure 1: Size and diversity of ExAC dataset a, ExAC dataset is almost ten fold bigger than datasets of similar kind: 1000 Genomes project and Exome Sequencing Project (ESP), but more importantly, it captures a far greater diversity of human populations compared to ESP and 1000 Genomes. b, The geographic signal of populations visualized using Principal component analysis (PCA). The first principal component get all the variability of African samples and it does not tells much about the rest of the dataset (Extended Data Figure 5 in the paper), therefore the second and third principal component has been show.
A 45 million nucleotide positions with sufficient coverage (>10x in at least 80% of individuals) are present in ExAC. These positions correspond to 18 million possible synonymous variants (in theory) of which ExAC is capturing 1.4 million (7.5%).
The size of ExAC allows to observe…
…mutational reoccurence: 43% of synonymous de Novo variants identified in previous studies were also identified in ExAC, which is a first direct evidence of mutational reoocuarence.
…multiple allels: 7.9% of high quality polymorphic sites are multiallelic, which is fairly close to Poisson expectation (whatever it means…)
…a LOT of variants after all the filtering, 7,404,909 high-quality variants were identified of which 317,381 indels. The density of variant is on the average one over eight bases. 99% of the variants had frequency bellow 1% and 54% of the variants are singletons (i.e. only one individual carries the variant).
…a selection effects The proportion of singletons among polymorphisms can serve as a measure of purifying selection acting on the polymorphisms of given size. The Figure 2 shows that indels that are not affecting open reading frame (ORF) have significantly less singleton variants than indels that actually affect ORF. There is also significant difference between indels of different sizes that are affecting ORF, but we (our topic group) have not found any possible explanation for this pattern.
…saturation of alleles in CpG sites: CpG sites have very high rate of transitions, therefore capturing all possible variants is substantially easier than for other sites. A subset of 20,000 individuals of ExAC dataset shows saturation of alleles – all non-lethal possible synonymous CpG transition variants are present. ExAC is the first dataset showing a saturation of human variation.
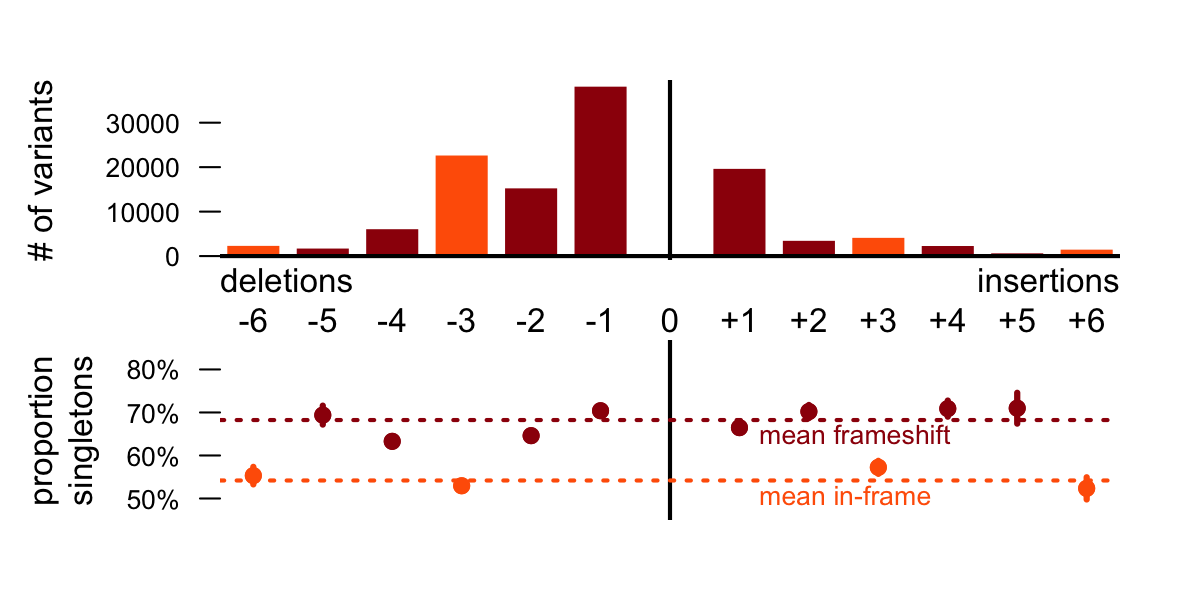
Figure 2: Indel frequencies with respect to the size a, Frequency of deletions is higher and smaller indels are more probable than greater. If we take into account the greater probability of smaller indels, frequency of indels that not shifting open reading frame is bit higher than frequency of indels than are not. b, Proportion of singletons in total number of indels (as proxy for strength of selection) is significantly and consistently lower in all indels that are not shifting open reading frame (-6, -3, +3, +6).
Deletireous alleles
Authors introduce a mutability adjusted proportion singleton (MAPS) metric as a measure of selection. This metric is correcting on biases caused by the different mutational rates allowing comparisons of categories with various mutational speed. Comparison across different functional classes have shown at Figure 3. MAPS shows higher values for categories predicted to be deleterious by conservation-based methods.
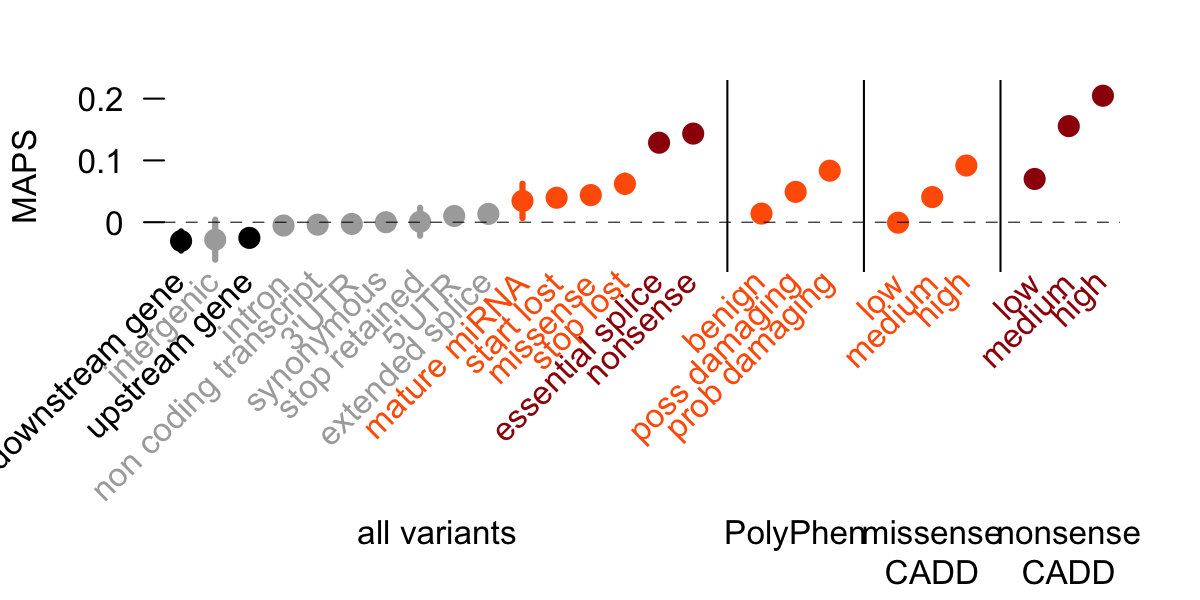
Figure 3: MAPS values of different functional classes. MAPS is highest for nonense substiturions and it also consistent with PolyPhen and Combined Annotation Dependent Depletion (CADD) classification.
Rare diseases
Average ExAC individual carries ~54 variants reported as Mendelian disease causing. Approximately 41 of these alleles were identified with frequency greater than one, therefore it is not expected to be caused by problem is variant calling, but in miss-classification of variants in the database. Evidence of 192 previously variants were manually curated of those only 9 had sufficient evidence in disease association. High allele frequencies were identified mainly in previously underrepresented categories Latino and South Asian.
ExAC have shown importance of matching reference population in identification disease-causing variant. An example is recessive disease North American Indian childhood cirrhosis previously reported to be caused by CIRH1A p.R565W. This variant was identified in homozygotic state in four individuals in Latino population, none of them having a record of liver problems during childhood.
Conclusion
ExAC shows the importance of diversity of sampled population in capturing the real link between genotype and phenotype. Even ExAC provides a lot of new insights, there are still populations that are underrepresented or not represented at all.
Given the richness of ExAC and the effort of authors in data sharing and availability, I guess that it will be a great resource for various analyses in the future for a lot of researchers around the globe.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, & Exome Aggregation Consortium. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536 (7616), 285-91 PMID: 27535533
One Comment on “ExAC presents a catalogue of human protein-coding genetic variation”
Comments are closed.
Separately, Walsh and colleagues from Imperial College London and the University of Oxford, U.K., have demonstrated the utility of the amassed genetic information for evaluating genes involved in multigenic heritable diseases. The team compared ExAC data to 7,855 clinical cardiomyopathy cases, finding that many putative cardiomyopathy-linked genes are unlikely to contribute to the disease. By focusing on confirmed genes, we expect this study to improve the clinical genetic testing of cardiomyopathies by reducing the number of uncertain and even false positive results, wrote Walsh. The team s results appeared in